Overview
The package mixhvg works for highly variable gene selection, including popular public available methods, and also the mixture of multiple highly variable gene selection methods. The mixture of methods can combine the advantages captured by each single method.
This function FindVariableFeaturesMix inherits from FindVariableFeatures function of Seurat Package, which can be used the same as FindVariableFeatures. Also, it accepts the dense or sparse matrix input.
Find the tutorial website here https://mixhvg.github.io.
By default, mixhvg accepts a raw count matrix as input and performs the necessary preprocessing internally, including log-normalization and PFlogPF transformation. However, users may also input a Seurat object. If the Seurat object contains only the raw count matrix, mixhvg will handle it the same way as a standalone count matrix. If the Seurat object includes both raw counts and user-processed log-normalized counts, then the log-normalized data in the Seurat object will be used directly for all methods that require it (e.g., lognc methods), while the raw counts will still be used for all other methods (e.g., ct, nc, and PFlogPF methods).
News
Sept. 15th, 2024: The package mixhvg is updated to version 1.0.1, which can support the use in Seurat version 5.
Sept. 1st, 2024: The package mixhvg is updated to version 0.2.1.
Aug. 26th, 2024: Our paper is on biorXiv as a preprint version: Zhao, R., Lu, J., Zhou, W., Zhao, N., & Ji, H. (2024). A systematic evaluation of highly variable gene selection methods for single-cell RNA-sequencing. bioRxiv, 2024-08.
Download
Please download the mixhvg package from CRAN.
install.packages("mixhvg")Please use GitHub repo to download the most updated package.
devtools::install_github("RuzhangZhao/mixhvg")Usage
There are two inputs can be used in FindVariableFeaturesMix function.
The example data comes from 10x Genomics. You may use this link to download the processed data. The processed data is named as pbmc3k_rna.rds.
- Matrix as input: only takes count matrix.
- Seurat Object as input:
- If log normalized result is in object: takes count and normalized matrices from Seurat Object.
- If log normalized result is not in object: takes count matrix from Seurat Object.
Seurat Object as Input
pbmc<-readRDS("pbmc3k_rna.rds")
library(Seurat)
library(mixhvg)
object<-CreateSeuratObject(pbmc)
object<-FindVariableFeaturesMix(object,nfeatures=2000)
head(VariableFeatures(object))
# [1] "CD74" "LYZ" "IGLC3" "IGKC" "RPS29" "IGHA1"One may use FindVariableFeaturesMix to replace the FindVariableFeatures function in the analysis pipeline of Seurat. For example,
object<-CreateSeuratObject(pbmc)
object<-FindVariableFeaturesMix(object)
object<-NormalizeData(object,verbose=FALSE)
object<-ScaleData(object,verbose = FALSE)
object<-RunPCA(object,npcs=30,verbose=FALSE)
object<-RunUMAP(object,dims=1:30,verbose = FALSE)Matrix as Input
pbmc<-readRDS("pbmc3k_rna.rds")
library(mixhvg)
pbmc_hvg<-FindVariableFeaturesMix(pbmc,nfeatures=2000)
head(pbmc_hvg)
# [1] "CD74" "LYZ" "IGLC3" "IGKC" "RPS29" "IGHA1"Different Methods
The method.names can take one method or multiple methods for mixture.
pbmc_hvg<-FindVariableFeaturesMix(pbmc,method.names="seuratv3")
pbmc_hvg<-FindVariableFeaturesMix(pbmc,method.names="scran")
pbmc_hvg<-FindVariableFeaturesMix(pbmc,
method.names=c("scran","scran_pos","seuratv1"))In the following example, we use a small cell sorting single-cell RNA-seq dataset with true label, downloaded from a large PBMC dataset. Download the example dataset using this link.
The data was provided by the work (Wang, J., Agarwal, D., Huang, M., Hu, G., Zhou, Z., Ye, C., & Zhang, N. R. (2019). Data denoising with transfer learning in single-cell transcriptomics. Nature methods, 16(9), 875-878.), which was initially downsampled from (Zheng, G. X., Terry, J. M., Belgrader, P., Ryvkin, P., Bent, Z. W., Wilson, R., … & Bielas, J. H. (2017). Massively parallel digital transcriptional profiling of single cells. Nature communications, 8(1), 14049.).
Load the scRNA-seq data and its corresponding labels.
expr<-readRDS("pbmc_900.rds")
cell_label<-read.table("cell_labels.txt",header = T)
cell_label<-factor(cell_label$cell_type_labels,levels = c("regulatory_t","naive_t","cd34","memory_t","cd56_nk","cytotoxic_t","cd14_monocytes","b_cells","naive_cytotoxic"))There are in total 9 cell type labels, with 100 cells for each cell type.
Here, we check the performance of two different HVG selection methods: Seurat v3 v.s. mixHVG. The evaluation is illustrated by the UMAP plots.
First, we show the standard pipeline for visualizing the dataset with default Seurat pipeline. Check Seurat tutorial for more details.
library(Seurat)
npcs=30
set.seed(1)
obj<-CreateSeuratObject(expr)
obj$cell_label<-cell_label
obj<-NormalizeData(obj,verbose=FALSE)
obj<-FindVariableFeatures(obj,verbose=FALSE)
obj<-ScaleData(obj,verbose=FALSE)
suppressWarnings(obj<-RunPCA(obj,npcs=npcs,verbose=FALSE))
obj<-RunUMAP(obj,dims = 1:npcs,verbose=FALSE)
pdf(paste0("umap_hvg_by_seuratv3.pdf"),height = 5.5,width = 7)
DimPlot(obj,reduction = "umap",group.by = "cell_label",shuffle = T)
dev.off() 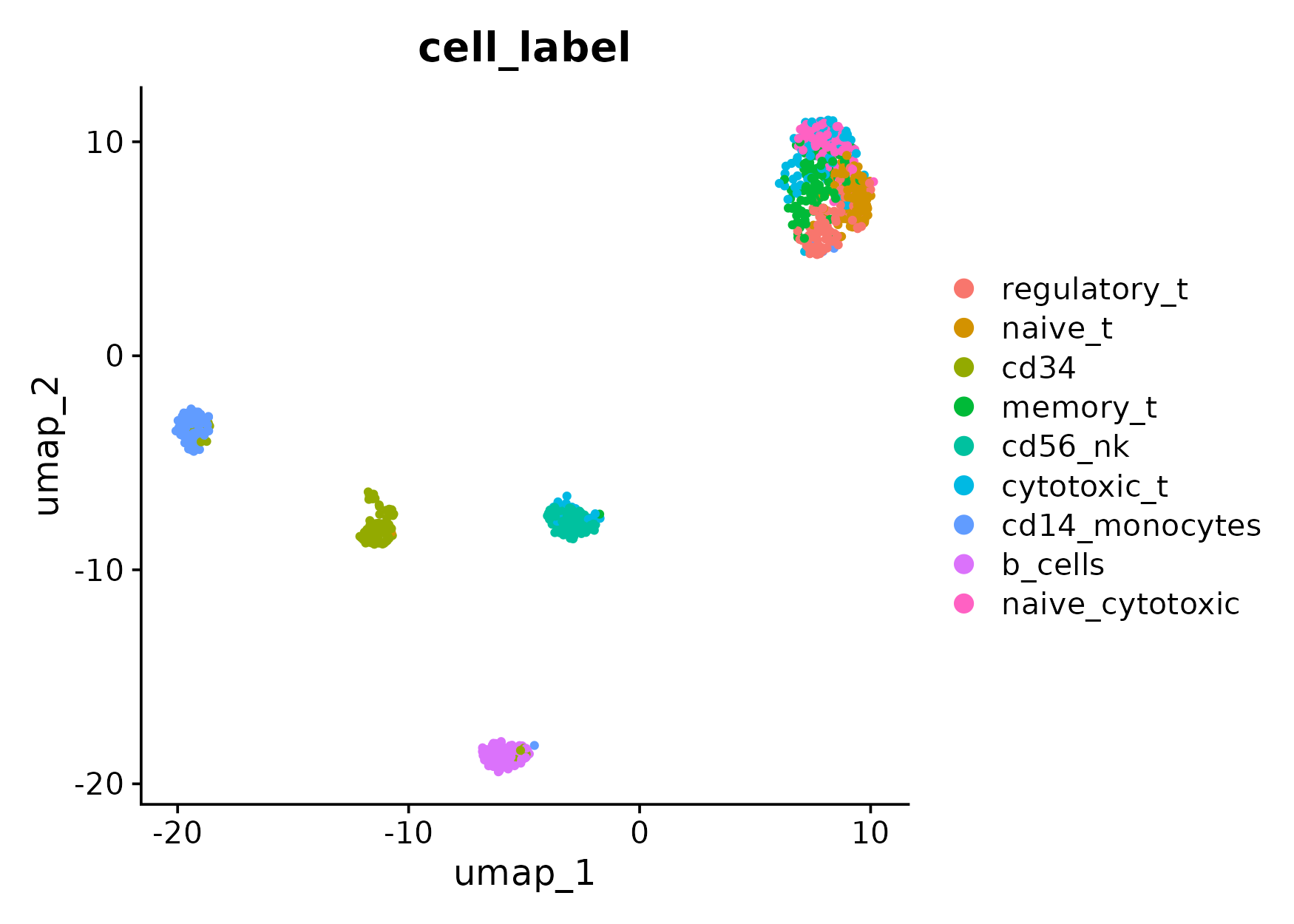
Second, we can simply replace the line FindVariableFeatures by FindVariableFeaturesMix with package mixhvg loaded to apply the mixHVG method.
library(Seurat)
library(mixhvg)
npcs=30
set.seed(1)
pbmc<-CreateSeuratObject(expr)
pbmc$cell_label<-cell_label
pbmc<-NormalizeData(pbmc,verbose=FALSE)
pbmc<-FindVariableFeaturesMix(pbmc,verbose=FALSE)
pbmc<-ScaleData(pbmc,verbose=FALSE)
suppressWarnings(pbmc<-RunPCA(pbmc,npcs=npcs,verbose=FALSE))
pbmc<-RunUMAP(pbmc,dims = 1:npcs,verbose=FALSE)
pdf(paste0("umap_hvg_by_mixhvg.pdf"),height = 5.5,width = 7)
DimPlot(pbmc,reduction = "umap",group.by = "cell_label",shuffle = T)
dev.off() 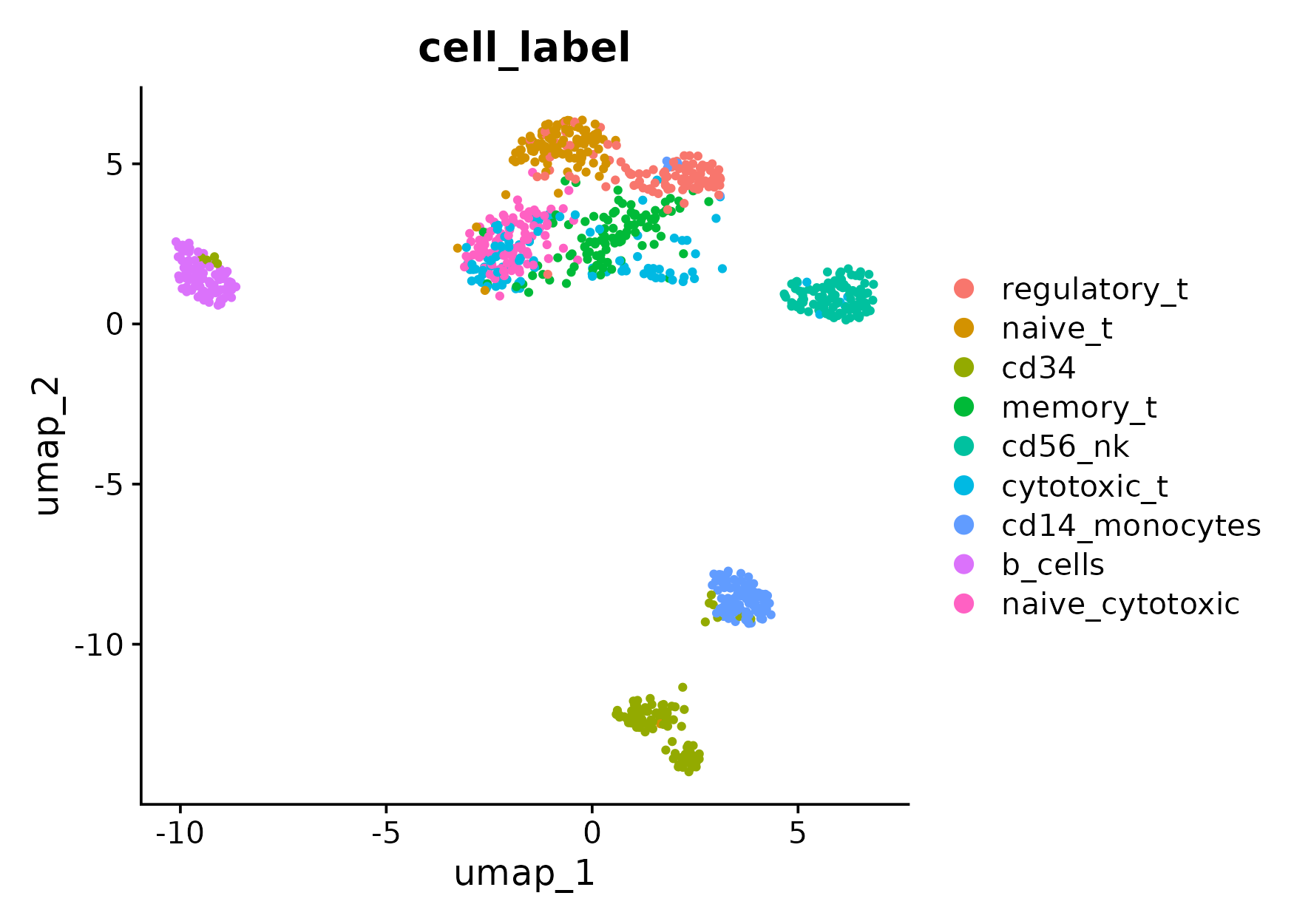
The evaluation is by adjusted rand index (ARI). Refer to our paper for more details.
maxARI<-function(embedding,label){
label = as.numeric(as.factor(label))
snn_<- FindNeighbors(object = embedding,
nn.method = "rann",
verbose = F)$snn
res<-sapply( seq(0,2,0.1) ,function(cur){
cluster_label <- FindClusters(snn_,resolution = cur,
verbose = F)[[1]]
mclust::adjustedRandIndex(cluster_label,label)})
max(res)
}
ari_seuratv3<-maxARI(obj@reductions$umap@cell.embeddings,cell_label)
ari_mixhvg<-maxARI(pbmc@reductions$umap@cell.embeddings,cell_label)ARI for UMAP with HVG selection by Seurat v3 is 0.6348
ARI for UMAP with HVG selection by mixHVG is 0.6767
Method Choices
The following methods can be chosen. And also, any mixture of the following methods is acceptable. For example, the default is c(“scran”,“seuratv1”,“mv_PFlogPF”,“scran_pos”)
- scran: Use mean-variance curve adjustment on lognormalized count matrix, which is scran ModelGeneVar.
- mv_ct: Use mean-variance curve adjustment on count matrix, inherited from scran ModelGeneVar.
- mv_nc: Use mean-variance curve adjustment on normalized count matrix, inherited from scran ModelGeneVar.
- mv_lognc: The same as scran.
- mv_PFlogPF: Use mean-variance curve adjustment on PFlog1pPF matrix, inherited from scran ModelGeneVar.
- scran_pos: Use scran poisson version, modelGeneVarByPoisson.
- seuratv3: Use logmean-logvariance curve adjustment on count matrix, which is vst, Seurat FindVariableFeatures Function(https://satijalab.org/seurat/reference/findvariablefeatures).
- logmv_ct: The same as seuratv3.
- logmv_nc: Use logmean-logvariance curve adjustment on normalized count matrix, inherited from seuratv3(vst).
- logmv_lognc: Use logmean-logvariance curve adjustment on lognormalized count matrix, inherited from seuratv3(vst).
- logmv_PFlogPF: Use logmean-logvariance curve adjustment on PFlog1pPF matrix, inherited from seuratv3(vst).
- seuratv1: Use dispersion on lognormalized count matrix, which is dispersion (disp), Seurat FindVariableFeatures Function(https://satijalab.org/seurat/reference/findvariablefeatures).
- disp_lognc: The same as seuratv1.
- disp_PFlogPF: Use dispersion on PFlog1pPF matrix, inherited from seuratv1(disp).
- mean_max_ct: Highly Expressed Features with respect to count matrix.
- mean_max_nc: Highly Expressed Features with respect to normalized count matrix.
- mean_max_lognc: Highly Expressed Features with respect to lognormalized count matrix
Appendix:
We can also include HIG as a baseline method in our mixture. HIG is a part of scCAD, a method designed for rare cell type identification. Please refer to our paper for more details.
For this purpose, we first install the scCAD Python package following the instructions on https://github.com/xuyp-csu/scCAD. According to the instructions, we will create a conda environment named scCAD_env, and install scCAD into this environment.
scCAD outputs a mixture of HVGs and HIGs, where the HVGs are identified by Seurat v1. We have modifed its source code to output only the HIGs. The modified code is provided in myscCAD.py. Please copy this file and paste it in the directory of the scCAD_env environment.
We will also need the reticulate R package to call Python from R. It can be installed and loaded using the following code.
if (!requireNamespace("reticulate", quietly = TRUE)) { install.packages("reticulate")}library(reticulate)Now we are ready to incorporate HIG in our mixture. This will be done in two steps: (1) We apply the HIG method and obtain the importance scores of the genes. We denote the ranking of the importance scores as extra.rank. (2) We use the FindVariableFeaturesMix function again, while using extra.rank as an input to this function.
We first apply HIG to our 900-cell example from the PBMC dataset.
use_condaenv("scCAD_env", required = TRUE)
scCAD <- import("scCAD")
np = import("numpy")
py_float <- py_eval("float")
data <- np$vectorize(py_float)(t(as.matrix(expr)))
geneNames = np$array(rownames(expr))
cellNames = np$array(colnames(expr))
HIGres = scCAD$myscCAD$getHIG(data=data,
dataName=dataset_name,
cellNames=cellNames, geneNames=geneNames)
selected_gene_names = HIGres[[1]]
selected_gene_imp = HIGres[[2]]
imp = rep(0, length(geneNames))
names(imp) = geneNames
imp[selected_gene_names] = selected_gene_imp
set.seed(1)
extra.rank = rank(-imp, ties.method = "random")
names(extra.rank) = gsub("_", "-", names(extra.rank))Then we call the FindVariableFeaturesMix and use extra.rank as an argument.
pbmc_with_HIG <- FindVariableFeaturesMix(pbmc, extra.rank=extra.rank, verbose=FALSE)